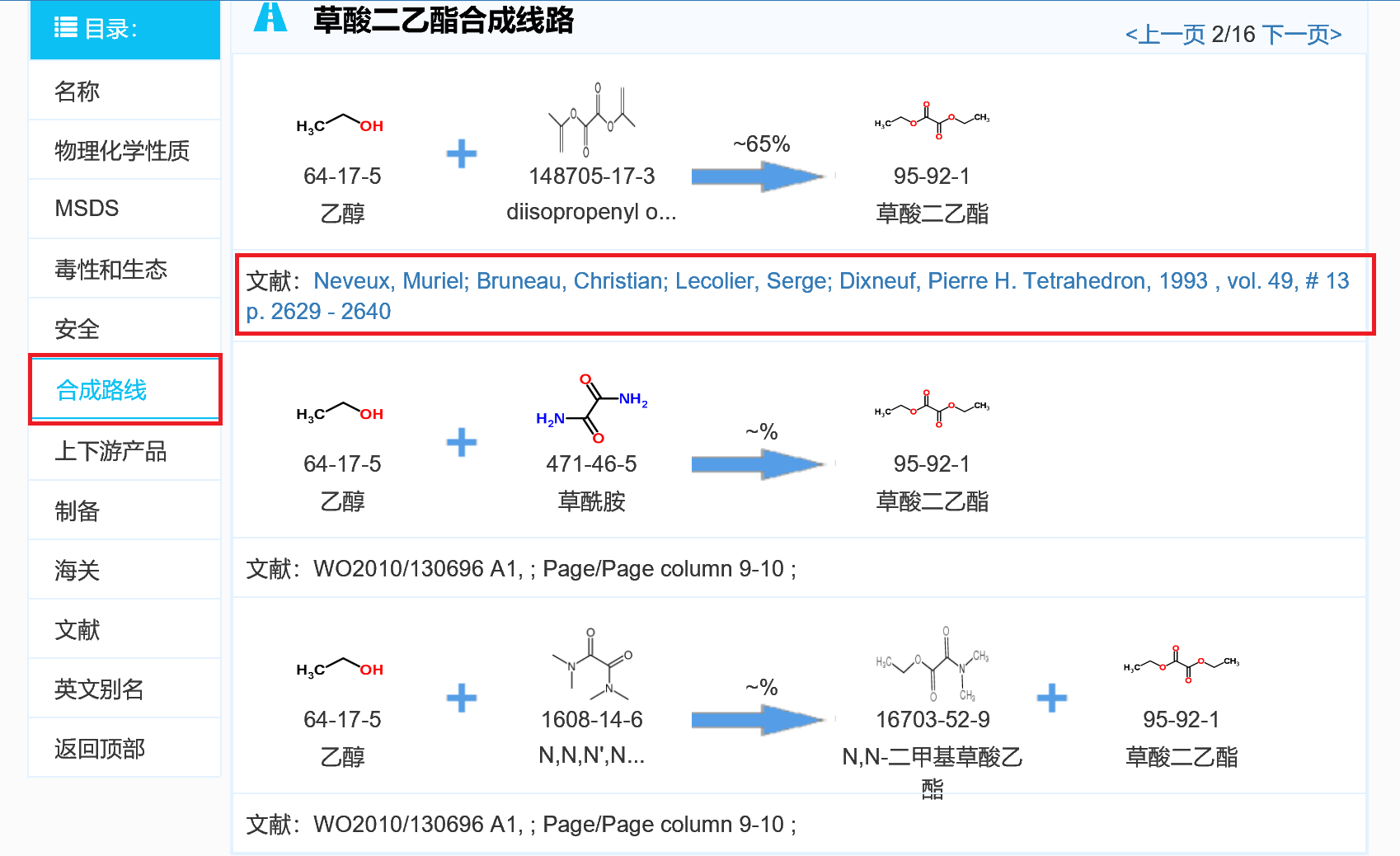
怎么使用ncbi的entrez检索工具
的有关信息介绍如下:
集成信息检索:ENTREZ系统编辑本段回目录检索服务器可以对有目标的检索记录,但它主要的缺陷在于一次只能从一个数据库中检索到记录;想对一批数据库进行检索的用户必须为每一个目标数据库分别发出一次申请。很明显,这些大量的公用数据库之间存在着逻辑联系。例如,MEDLINE中的一篇论文可能描述一个基因的序列,该基因又在GenBank中出现。其核苷酸序列所编码的蛋白质的序列又存放在蛋白质数据库中。这种蛋白质的三维结构可能又是已知的,结构的数据可能出现在结构数据库中。最后,基因可能定位在某条染色体的某个区域,这类信息存放在图谱数据库中。在这些生物学上的联系的基础上开发了一种方法;可以通过它查询所有与某一特殊的生物学实体有关的所有信息,而不必按次序查询分立的数据库。这就是一个名为Entrez的分子检索系统。它由NCBI开发和维护,Entrez在所有的主要的数据库计算机平台上均可使用,允许对PubMed(MEDINE)的记录,核苷酸和蛋白质的序列数据,三维结构信息,图谱信息进行集成的访问。全部信息只需经过一次查询。Entrez能够通过数据库之间的两种类型联系:相近性和硬连接来提供集成的信息检索。相近性相近性联系着一个给定的数据库之内的记录。使用者在查看MEDLINE中某条记录时可以要求Entrez"找出所有类似的论文",类似的,使用者在查看一个序列的同时可以要求Entrez"找出所有与这个序列类似的序列"。一个数据库之内的相近性关系是建立在对相似性的统计计算上的:BLAST 序列数据可以用基本局部对比搜索工具(Basic Local Alignment Search Tool,即BLAST)相互比较。这个算法试图找到"高度匹配的片段对"(high-scoring segment pairs,简记为HSPs),即能够无缺口的对齐且达到一定的分数的成对的序列。VAST 几套坐标数据之间的比较采用一种名为VAST的基于向量的算法。VAST即Vector Alignment Search Tool(Madej等,1995;Gibrat等,1996)。VAST的比较有三个步骤:1.第一,在坐标数据的基础上,标出所有的构成蛋白质的核心部分的α螺旋和β片层。然后根据这些二级结构单位的位置计算向量。以下的步骤使用这些向量来做对比而不是整个一套坐标。2.然后,算法试图最佳的匹配这些向量,寻找类型和相对方位相同的成对的结构单位,并且在这些单位之间还要有同样的连接方式。其目标在于识别高度相似的"核心结构",这些成对结构的匹配性要比随机的选择蛋白质相互比较得到的高得多。3.最后,在每个残基位置上使用蒙特-卡洛方法对结构的排列进行优化。使用这个方法有可能找到一些序列相似性不明显的蛋白质之间的结构上的关系(可能在功能上也有关系)。最后的对齐结果不一定是全局的,可能在不同的蛋白质的单独的结构域之间配对。需要重点注意的是VAST不是确定结构相似性的最好办法,因为还可以利用三维坐标文件中的其它信息来做更进一步的修正,如考虑侧链的位置及侧链之间的相互作用的热力学特点。而把结构压缩成一列矢量必然会导致信息的丢失。然而,考虑到这个问题的数量级-即需要做的成对比较的次数-及采用更高级的方法所需要的计算能力和时间,VAST至少为结构相似性问题提供了一个简单和快速的答案。